In [113]:
# Plot the first two dimensions
import matplotlib.pyplot as plt
plt.scatter(uce[:, 0], uce[:, 1], s=1)
plt.xlabel("Dimension 1")
plt.ylabel("Dimension 2")
plt.title("UCE Embedding")
plt.show()

Tyler Burns
October 12 - October 13, 2024
This markdown looks at the output of the PBMC 3k and PBMC 10k datasets when run through the Universal Cell Embeddings foundation model.
The spoiler alert is I initially think the two datasets are going to sit on top of each other because the transformer will have "grokked" integration, but I find out that isn't the case.
However, I find using my tool KNN sleepwalk, that the distances in UMAP space don't capture the distanecs in the UCE manifold, where the corresponding subsets within each dataset are actually quite closer to each other (in other words, the T cells in the PBMC 3k dataset are sitting right next to the T cells in the PBMC 10k dataset, but this is not reflected on the UMAP).
We start with the PBMC 10k dataset.
import scanpy as sc
import random
random.seed(1)
import warnings
warnings.filterwarnings('ignore')
pb10k = sc.read_h5ad("../output/33_layer/10k_pbmcs_proc_uce_adata.h5ad")
pb10k
AnnData object with n_obs × n_vars = 11990 × 10809
obs: 'n_counts', 'batch', 'labels', 'str_labels', 'cell_type', 'n_genes'
var: 'gene_symbols', 'n_counts-0', 'n_counts-1', 'n_counts', 'highly_variable', 'highly_variable_rank', 'means', 'variances', 'variances_norm', 'n_cells'
uns: 'cell_types', 'hvg'
obsm: 'X_uce', 'design', 'normalized_qc', 'qc_pc', 'raw_qc'
Ok, now we've got the cells, and we can have a look at the embeddings. We will go right into it.
# Pull out X_uce from obsm
uce = pb10k.obsm["X_uce"]
uce.shape
(11990, 1280)
As a sanity check, we see 1280 dimensions that the UCE gave us, for just over 10k cells. Now we plot the first two dimensions.
# Plot the first two dimensions
import matplotlib.pyplot as plt
plt.scatter(uce[:, 0], uce[:, 1], s=1)
plt.xlabel("Dimension 1")
plt.ylabel("Dimension 2")
plt.title("UCE Embedding")
plt.show()
Ok, so unlike PCA, we don't get any interesting information from plotting two dimensions out of the 1280, it seems. Time to run UMAP.
import umap
# Run UMAP on the embedding
reducer = umap.UMAP()
embedding = reducer.fit_transform(uce)
# Plot it
plt.scatter(embedding[:, 0], embedding[:, 1], s=1)
plt.xlabel("UMAP 1")
plt.ylabel("UMAP 2")
plt.title("UMAP Embedding")
plt.show()

So now we have a sanity check. The PBMCs should break into around 3 islands. This is generally how they look with scRNA seq data.
However, the interesting finding here is that the model was given raw counts. There was no processing (eg. finding the most variable genes, taking the top n PCs of that, etc). And we got a UMAP that looks like the data were preprocessed. So the foundation model is doing something in the right direction. That is pretty cool.
Now we're going to color the umap by cell type. First let's look at the metadata.
# Show metadata in the anndata object
pb10k.obs.head()
| n_counts | batch | labels | str_labels | cell_type | n_genes | |
|---|---|---|---|---|---|---|
| AAACCTGAGCTAGTGG-1 | 4520.0 | 0 | 2 | CD4 T cells | CD4 T cells | 735 |
| AAACCTGCACATTAGC-1 | 2788.0 | 0 | 2 | CD4 T cells | CD4 T cells | 449 |
| AAACCTGCACTGTTAG-1 | 4667.0 | 0 | 1 | CD14+ Monocytes | CD14+ Monocytes | 942 |
| AAACCTGCATAGTAAG-1 | 4440.0 | 0 | 1 | CD14+ Monocytes | CD14+ Monocytes | 924 |
| AAACCTGCATGAACCT-1 | 3224.0 | 0 | 3 | CD8 T cells | CD8 T cells | 691 |
We find that the cells are indeed annotated. It looks like labels, str_labels, and cell_type are redundant. But let's see for ourselves.
# Check to see if the metadata columns str_labels and cell_type are redundant
(pb10k.obs["str_labels"] == pb10k.obs["cell_type"]).all()
np.True_
Yes. Now let's color the UMAP by cell type.
# Use scanpy to plot the UMAP
pb10k.obsm['X_umap'] = embedding
sc.pl.umap(pb10k, color='cell_type')

Ok, so its producing data that passes my sanity check. This is good.
Now we are going to upload the PBMC 3k dataset and check it. After that, we're going to combine the datasets and see if they "integrate."
# Upload the PBMC 3k dataset
pb3k = sc.read_h5ad("../output/33_layer/pbmc3k_raw_uce_adata.h5ad")
pb3k
AnnData object with n_obs × n_vars = 2700 × 9714
obs: 'n_genes'
var: 'gene_ids', 'n_cells'
obsm: 'X_uce'
Now we're going to upload the "processed" dataset (available through scanpy) so we can get the cell subsets. This processed dataset returned an error when fed into the model, so we ran the "raw" dataset through the transformer (available through scanpy), and we now have to get the relevant data from the "processed" dataset here.
# Read in the data
pb3k_proc = sc.read_h5ad("../output/4_layer/pbmc3k_processed_proc.h5ad")
cell_type = pb3k_proc.obs["louvain"]
cell_type
index
AAACATACAACCAC-1 CD4 T cells
AAACATTGAGCTAC-1 B cells
AAACATTGATCAGC-1 CD4 T cells
AAACCGTGCTTCCG-1 CD14+ Monocytes
AAACCGTGTATGCG-1 NK cells
...
TTTCGAACTCTCAT-1 CD14+ Monocytes
TTTCTACTGAGGCA-1 B cells
TTTCTACTTCCTCG-1 B cells
TTTGCATGAGAGGC-1 B cells
TTTGCATGCCTCAC-1 CD4 T cells
Name: louvain, Length: 2638, dtype: category
Categories (8, object): ['CD4 T cells', 'CD14+ Monocytes', 'B cells', 'CD8 T cells', 'NK cells', 'FCGR3A+ Monocytes', 'Dendritic cells', 'Megakaryocytes']
# Add cell_type to pb3k.obs, given the index for each, but don't name index
pb3k.obs["cell_type"] = cell_type
pb3k.obs
| n_genes | cell_type | |
|---|---|---|
| index | ||
| AAACATACAACCAC-1 | 778 | CD4 T cells |
| AAACATTGAGCTAC-1 | 1346 | B cells |
| AAACATTGATCAGC-1 | 1126 | CD4 T cells |
| AAACCGTGCTTCCG-1 | 953 | CD14+ Monocytes |
| AAACCGTGTATGCG-1 | 520 | NK cells |
| ... | ... | ... |
| TTTCGAACTCTCAT-1 | 1148 | CD14+ Monocytes |
| TTTCTACTGAGGCA-1 | 1215 | B cells |
| TTTCTACTTCCTCG-1 | 618 | B cells |
| TTTGCATGAGAGGC-1 | 449 | B cells |
| TTTGCATGCCTCAC-1 | 722 | CD4 T cells |
2700 rows × 2 columns
# Run umap on the PBMC 3k dataset
pb3k_embedding = reducer.fit_transform(pb3k.obsm["X_uce"])
# Add umap to the obs of the pb3k dataset
pb3k.obsm["X_umap"] = pb3k_embedding
# Use scanpy umap plot
sc.pl.umap(pb3k, color='cell_type', title='PBMC 3k UMAP colored by cell type')

Similar shape, and the cell types shake out as I expect them to. So it passes my sanity check.
Now for the question as to whether UCE can grok integration. Will the datasets sit on top of each other when I run UMAP on the concatenated PBMC 3k + PBMC 10k dataset? Let's find out.
# Combine the two datasets, making sure they have distinct names in the metadata
pb10k.obs["dataset"] = "10k"
pb3k.obs["dataset"] = "3k"
com = pb10k.concatenate(pb3k)
com
AnnData object with n_obs × n_vars = 14690 × 8235
obs: 'n_counts', 'batch', 'labels', 'str_labels', 'cell_type', 'n_genes', 'dataset'
var: 'gene_symbols-0', 'n_counts-0-0', 'n_counts-1-0', 'n_counts-0', 'highly_variable-0', 'highly_variable_rank-0', 'means-0', 'variances-0', 'variances_norm-0', 'n_cells-0', 'n_cells-1', 'gene_ids-1'
obsm: 'X_uce', 'X_umap'
com.obs
| n_counts | batch | labels | str_labels | cell_type | n_genes | dataset | |
|---|---|---|---|---|---|---|---|
| AAACCTGAGCTAGTGG-1-0 | 4520.0 | 0 | 2.0 | CD4 T cells | CD4 T cells | 735 | 10k |
| AAACCTGCACATTAGC-1-0 | 2788.0 | 0 | 2.0 | CD4 T cells | CD4 T cells | 449 | 10k |
| AAACCTGCACTGTTAG-1-0 | 4667.0 | 0 | 1.0 | CD14+ Monocytes | CD14+ Monocytes | 942 | 10k |
| AAACCTGCATAGTAAG-1-0 | 4440.0 | 0 | 1.0 | CD14+ Monocytes | CD14+ Monocytes | 924 | 10k |
| AAACCTGCATGAACCT-1-0 | 3224.0 | 0 | 3.0 | CD8 T cells | CD8 T cells | 691 | 10k |
| ... | ... | ... | ... | ... | ... | ... | ... |
| TTTCGAACTCTCAT-1-1 | NaN | 1 | NaN | NaN | CD14+ Monocytes | 1148 | 3k |
| TTTCTACTGAGGCA-1-1 | NaN | 1 | NaN | NaN | B cells | 1215 | 3k |
| TTTCTACTTCCTCG-1-1 | NaN | 1 | NaN | NaN | B cells | 618 | 3k |
| TTTGCATGAGAGGC-1-1 | NaN | 1 | NaN | NaN | B cells | 449 | 3k |
| TTTGCATGCCTCAC-1-1 | NaN | 1 | NaN | NaN | CD4 T cells | 722 | 3k |
14690 rows × 7 columns
Now we make the UMAP and color by datasets to see where they sit relative to each other.
# Make a umap from the combined data
com_uce = com.obsm["X_uce"]
com_reducer = umap.UMAP(random_state=1) # Good states: 1, 5, 6
com_embedding = com_reducer.fit_transform(com_uce)
com.obsm["X_umap"] = com_embedding
sc.pl.umap(com, color='dataset', title='Combined PBMC UMAP colored by dataset')

And it turns out that they are separate, though they seem to line up. UCE didn't fully grok integration, but let's investigate further by coloring by cell type.
sc.pl.umap(com, color='cell_type', title='Combined UMAP colored by cell type')

We see that there is some sort of "axis" in which the cell types for each dataset line up. This is interesting. We are going to output the data and investigate this by looking at the k-nearest neighbors in UCE space, which will tell us whether UMAP's distance is off, and the cell types corresponding to each dataset are sitting right next to each other.
We will use KnnSleepwalk, my fork of the sleepwalk package, which I use to visualize the KNN of the original high-dimensional manifold that UMAP compresses into two dimensions.
So we'll output the anndata below so we can get this into R.
# Save the combined anndata object
com.write_h5ad("../output/33_layer/combined_pbmc_uce_adata.h5ad")
We run KnnSleepwalk in R. Look again at the UMAPs above colored by dataset. Now have a look at this labeled gif:
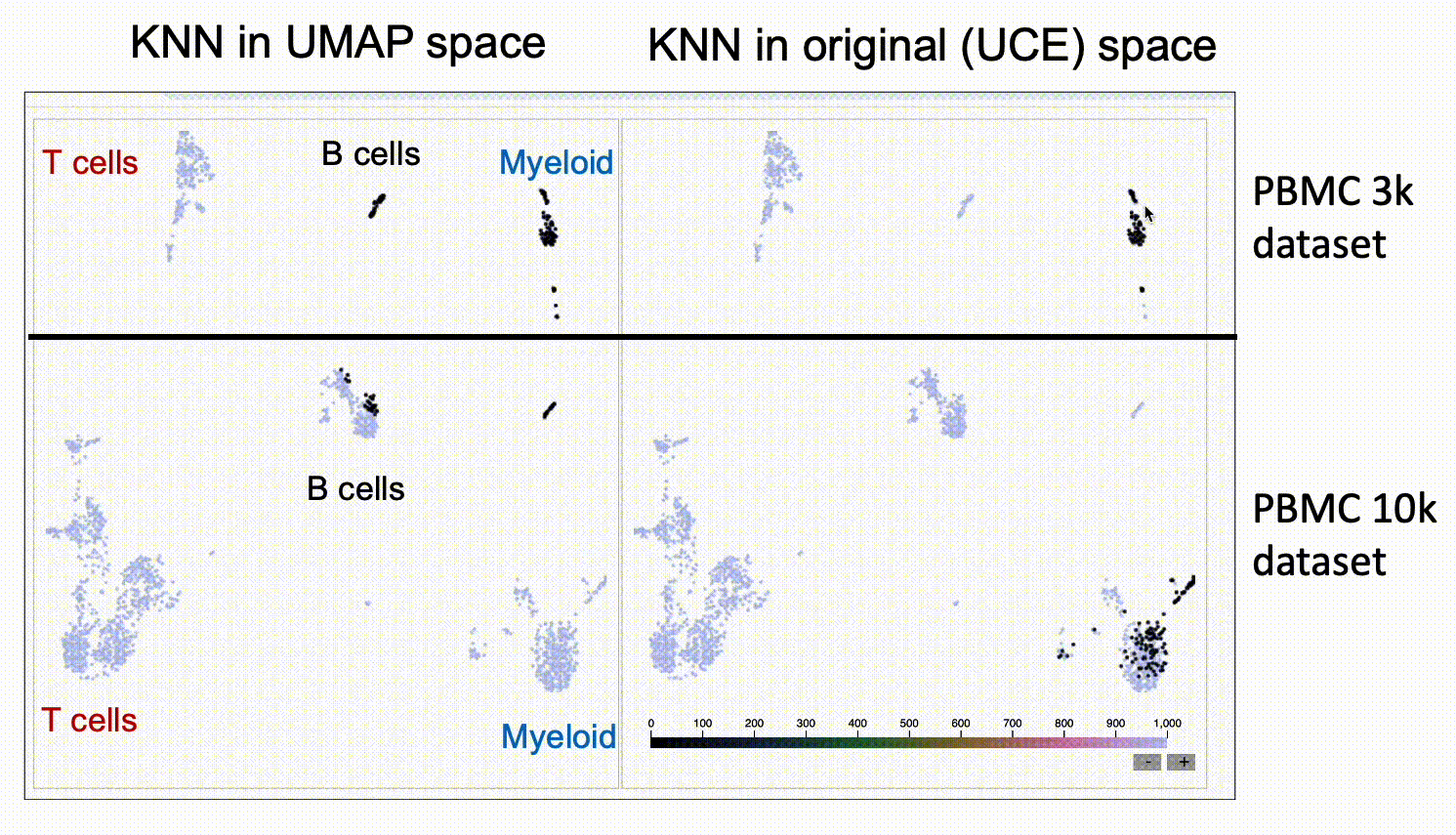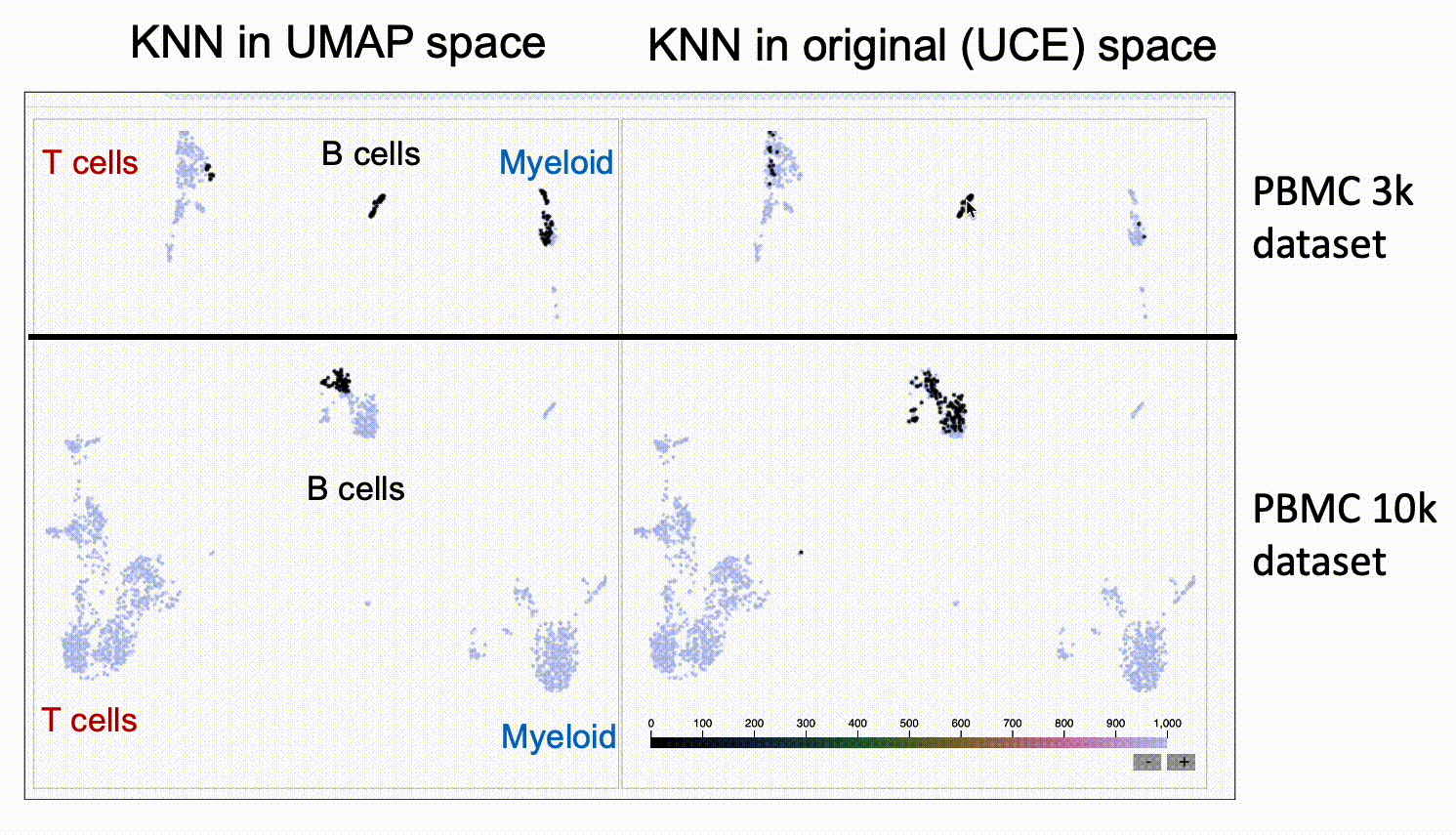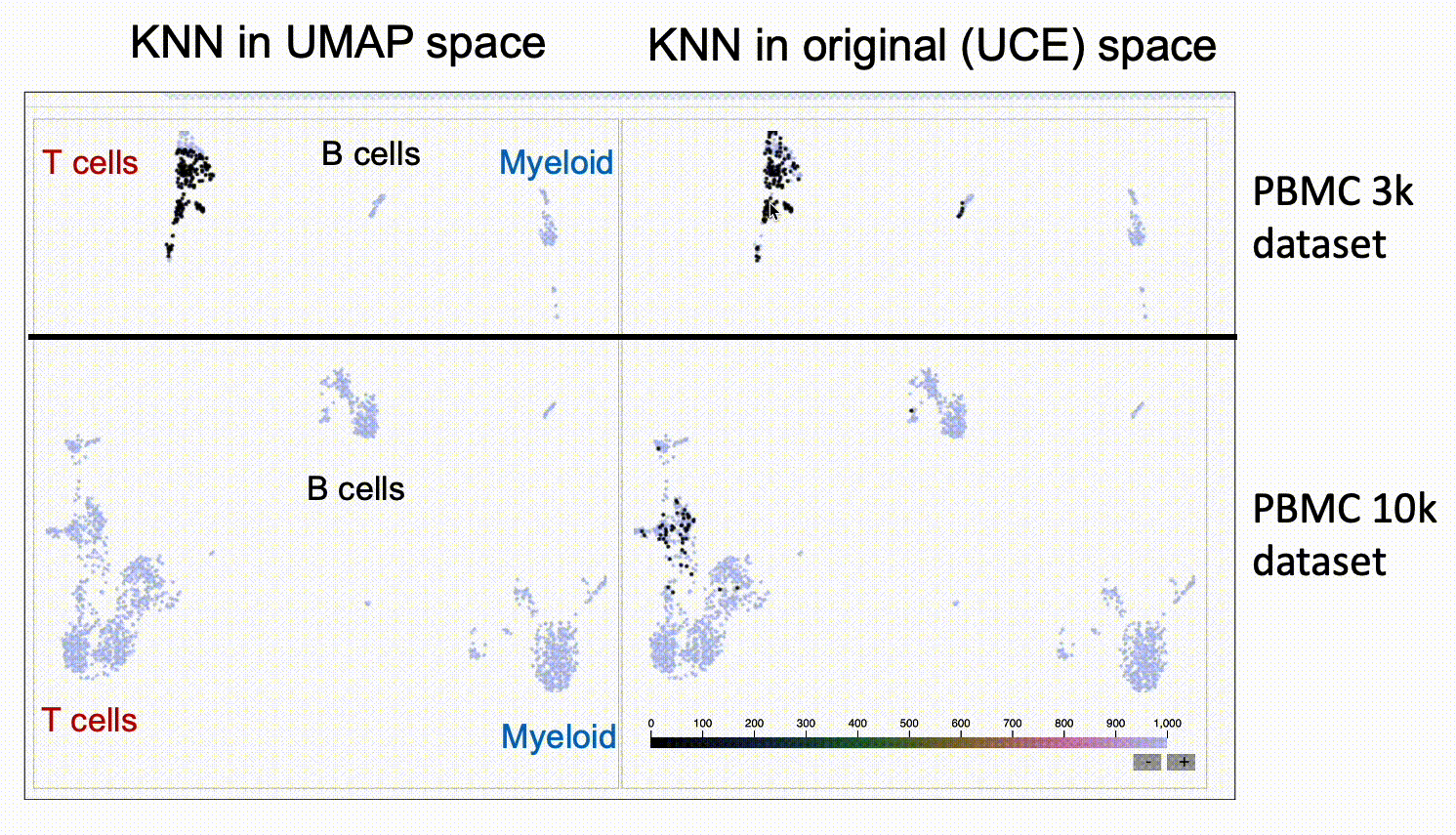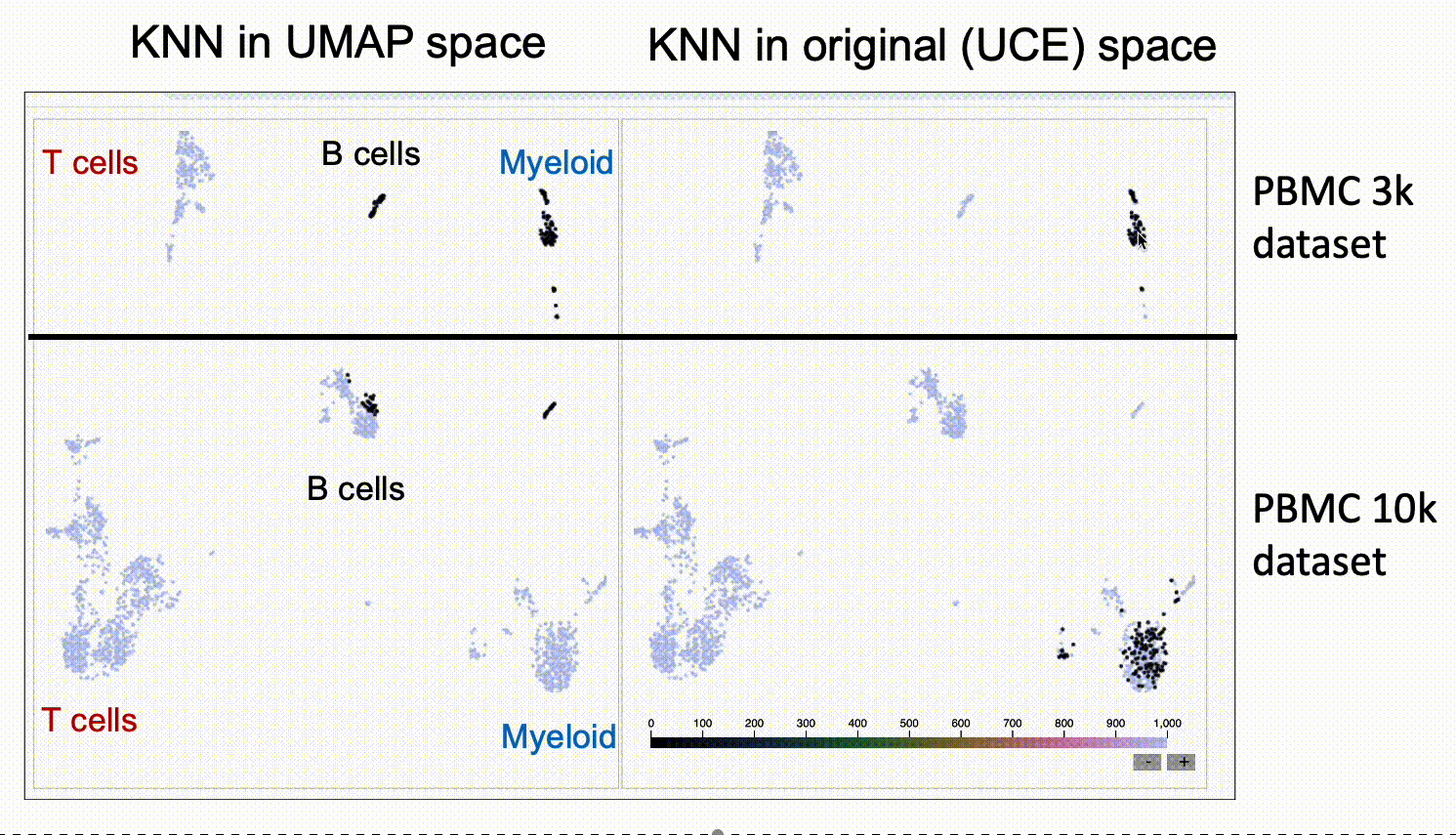
Then, if you want to play around with this yourself, please go to this link.
The UMAP on the left will give you the nearest neighbors to the cursor in UMAP space. The UMAP in the right will give you the nearest neighbors in the high-dimensional UCE space. You'll see that the one on the right has "out of island" nearest neighbors that correspond to the given cell type of the other dataset.
This suggests that UCE might indeed have the ability to at least partially grok integration, and that UMAP might not be the best way to assess it, as UMAP space seems to distort the "distances" between datasets.
The last thing I'll show is that the PCA space of the UCE seems to show a specific axis that corresponds to the dataset. In this case, it's PC2. In other words, if you take one dataset and "slide" it in the right direction along PC2, it will merge with the other dataset.
# Make PCA of com uce space without scanpy
from sklearn.decomposition import PCA
pca = PCA(n_components=50)
pca.fit(com_uce)
com_pca = pca.transform(com_uce)
# Plot the first two dimensions in scanpy
com.obsm["X_pca"] = com_pca
sc.pl.pca(com, color='dataset', title='Combined PCA colored by dataset')
# And plot by cell type
sc.pl.pca(com, color='cell_type', title='Combined PCA colored by cell type')


Taken together, UCE takes raw counts as input and produces a 1280 dimensional embedding that in turn produces UMAPs that seem to resemble what you'd otherwise get if you did the standard pre-processing (find variable genes, normalize, scale, first n principal components).
When 2 PBMC datasets were run through UCE, the UMAPs seemed to suggest that batch effects between the datasets were not "grokked" by the model. However, two things seem to suggest the contrary.
The first is that KNN sleepwalk shows that UMAP distorted the distances between the datasets. In other words, islands that correspond to particular cell types are largely "nearest neighbors" of the islands of the respective cell types in the other dataset.
The second is that the first two principal components of the UCE embedding show that PC2 largely corresponds to the datasets, such that you could slide one onto the other using just that axis.
Thus, UCE seems to position a cell type of one dataset near to a cell type of another dataset, but UMAP distorts the distances so it might not appear so.
In other words, when interrogating foundation models like UCE that output high-dimensional embeddings, one must be careful when using non-linear dimensionality reduction tools when characterizing them.